Huntington’s Disease
Huntington's disease is an autosomal-dominant, progressive neurodegenerative disorder with a distinct phenotype, including chorea and dystonia, incoordination, cognitive decline, and behavioural difficulties. Typically, onset of symptoms is in middle-age after affected individuals have had children, but the disorder can manifest at any time between infancy and senescence. The mutant protein in Huntington's disease—huntingtin—results from an expanded CAG repeat leading to a polyglutamine strand of variable length at the N-terminus.
The DNA error that causes HD is found in a gene called huntingtin. This gene was discovered in 1993. Everyone has the huntingtin gene, but only those that inherit the mistake, known as the HD mutation, will develop HD and risk passing it on to their children. Genes are made up of the nucleotide “letters” A,G,C, and T, which form a code that is read in groups of three. HD is caused by a stretch of the letters C-A-G in the huntingtin gene which repeat over and over, too many times…CAGCAGCAGCAGCAG. This is known as a CAG repeat expansion. In the huntingtin gene, most people have around 20 CAG repeats, but people with HD have around 40 or more. Every person who has this CAG repeat expansion in the HD gene will eventually develop the disease, and each of their children has a 50% chance of developing HD.
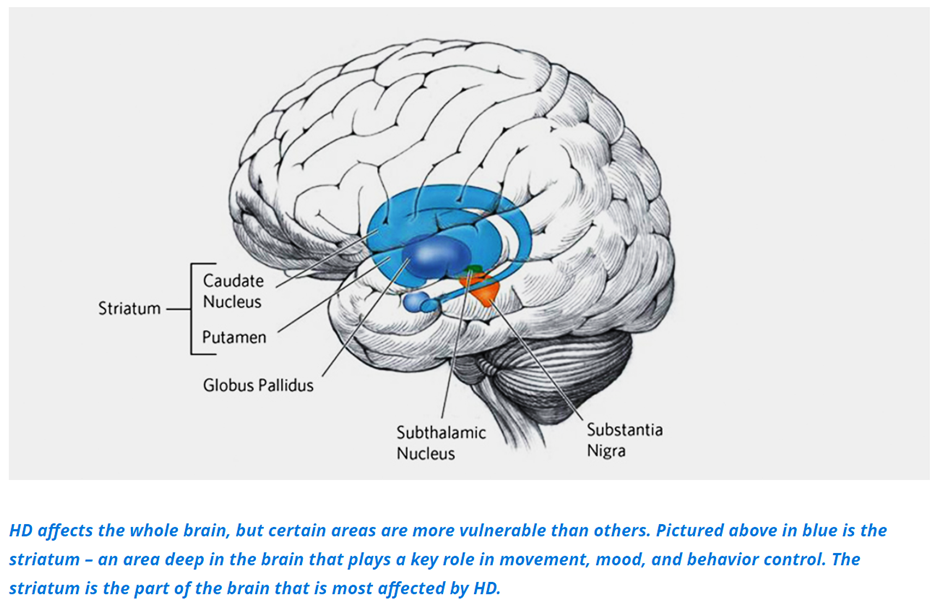
Our genes are like an instruction manual for making proteins, the machines that run everything in our bodies. The huntingtin gene (DNA) contains instructions that are copied into a biological message (RNA) which makes the huntingtin protein. The huntingtin protein is very large and seems to have many functions, especially as the brain is developing before birth, but it is not fully understood. We know that the extra CAG repeats in people with HD cause the huntingtin protein to be extra-long and difficult to maintain, which makes it difficult for it to do its job. Over many years, this “mutant” huntingtin protein forms clumps in brain cells, and causes them to become damaged and die. The most vulnerable part of the brain in HD is called the striatum, and it controls movement, mood, and memory. Damage to the striatum over time is what causes the symptoms of HD.
What are the symptoms?
Symptoms vary from person to person and at different stages of the disease.
- Physical symptoms: weight loss, involuntary movements (chorea), diminished coordination, difficulty walking, talking and swallowing
- Cognitive symptoms: difficulty with focus, planning, recall of information and making decisions; impaired insight
- Emotional symptoms: depression, apathy, irritability, anxiety, obsessive behaviour
Early Stages of HD
At this stage, people with HD can function quite well at work and at home. Early symptoms of the disease may include the following:
- Difficulty organizing routine matters or coping effectively with new situations
- Decreased ability to recall information and make decisions
- Increased difficulty with work activities
- Decreased attention to details
- Mood changes and irritability
- Minor involuntary movements (e.g. “nervous” activity, fidgeting, a twitching of the limbs or excessive restlessness)
- Changes in handwriting or difficulty with daily tasks such as driving
Intermediate stages of HD
People with HD in the intermediate stage may have increasing difficulty working or managing a household but can still deal with most activities of daily living. Symptoms progress over time and may include the following:
- More obvious involuntary movements (chorea)
- Increased difficulty with walking, coordination and balance
- Challenges with speaking (speech may become slurred) and delays in thinking process
- Solving problems becomes more difficult
- Difficulties with swallowing
- Weight loss
Advanced stages of HD
People in the advanced stages of HD can no longer manage the activities of daily living and usually require professional care. Symptoms will include the following:
- Decrease in involuntary movements and increase in rigidity
- Increased difficulties with swallowing
- The ability to communicate diminishes, but understanding what is being said remains possible
- Significant weight loss
Juvenile HD
About 10% of people diagnosed
with Huntington disease have the juvenile form. In Juvenile Huntington Disease
(JHD), the symptoms occur in childhood or adolescence (before the age of 20)
and tend to follow a more rapid course. Diagnosis of JHD is very difficult
because the symptoms of Juvenile HD have somewhat different features from the
adult form of the disease. Chorea (involuntary movements) is a much less
prominent feature and may be absent altogether.
Initial symptoms may include the following:
- Slow and stiff movement (rigidity), and sometimes tremors
- Difficulty learning in school and attention deficits
- Increase in responsive behaviours
- Seizures
Having a team of professionals to work with the person with JHD and the family is very important.
Late onset HD (diagnosis after age 60)
There is a wide range in the age of disease onset for people with HD. If a diagnosis is received after age 60, it is considered Late Onset HD. Knowledge of the typical age of onset (ages 35 to 55) sometimes leads physicians to miss the diagnosis, because doctors incorrectly believe the person is too old to develop HD. Late onset consists of about 10% of all HD diagnoses.
Causes
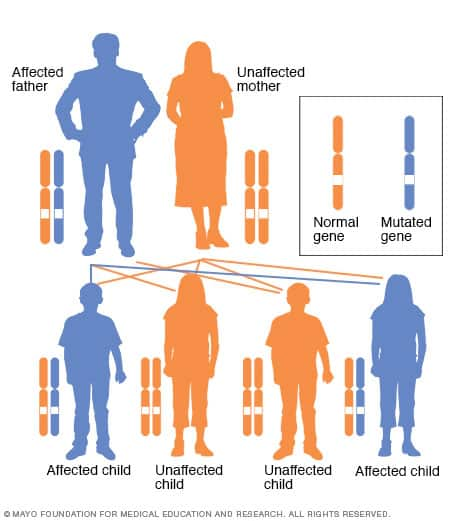
Autosomal dominant inheritance pattern
Huntington's disease is caused by an inherited difference in a single gene. Huntington's disease is an autosomal dominant disorder, which means that a person needs only one copy of the nontypical gene to develop the disorder.
With the exception of genes on the sex chromosomes, a person inherits two copies of every gene — one copy from each parent. A parent with a nontypical gene could pass along the nontypical copy of the gene or the healthy copy. Each child in the family, therefore, has a 50% chance of inheriting the gene that causes the genetic disorder.