INTRODUCTION
Thalassemia is a genetically inherited blood disorder which is characterized by the production of less or abnormal hemoglobin. As we know, hemoglobin is a protein found in Red Blood Cells. Hemoglobin is responsible for carrying oxygen around in the body. Thalassemia results in large numbers of red blood cells being destroyed, which leads to anemia. Anemia is a condition in which your body doesn’t have enough normal, healthy red blood cells. As a result of anemia, person affected with Thalassemia will have pale skin, fatigue and dark coloration of urine.
Thalassemia is inherited, meaning that at least one of your parents must be a carrier of the disorder. It’s caused by either a genetic mutation or a deletion of certain key gene fragments. Thalassemia minor is a less serious form of the disorder. There are two main forms of thalassemia that are more serious. In alpha thalassemia, at least one of the alpha globin genes has a mutation or abnormality. In beta thalassemia, the beta globin genes are affected.
Thalassemia beta
Beta thalassemia occurs when your body can’t produce beta globin. Two genes, one from each parent, are inherited to make beta globin. This type of thalassemia comes in two serious subtypes: thalassemia major (Cooley’s anemia) and thalassemia intermedia.
Thalassemia major
Thalassemia major is the most severe form of beta thalassemia. It develops when beta globin genes are missing.
The symptoms of thalassemia major generally appear before a child’s second birthday. The severe anemia related to this condition can be life-threatening. Other signs and symptoms include:
- fussiness
- paleness
- frequent infections
- a poor appetite
- failure to thrive
- jaundice, which is a yellowing of the skin or the whites of the eyes
- enlarged organs
This form of thalassemia is usually so severe that it requires regular blood transfusions.
Thalassemia intermedia
Thalassemia intermedia is a less severe form. It develops because of alterations in both beta globin genes. People with thalassemia intermedia don’t need blood transfusions.
Thalassemia alpha
Alpha thalassemia occurs when the body can’t make alpha globin. In order to make alpha globin, you need to have four genes, two from each parent.
This type of thalassemia also has two serious types: hemoglobin H disease and hydrops fetalis.
Hemoglobin H
Hemoglobin H develops as when a person is missing three alpha globin genes or experiences changes in these genes. This disease can lead to bone issues. The cheeks, forehead, and jaw may all overgrow. Additionally, hemoglobin H disease can cause:
- jaundice
- an extremely enlarged spleen
- malnourishment
Hydrops fetalis
Hydrops fetalis is an extremely severe form of thalassemia that occurs before birth. Most babies with this condition are either stillborn or die shortly after being born. This condition develops when all four alpha globin genes are altered or missing.
Thalassemia and anemia
Thalassemia can quickly lead to anemia. This condition is marked by a lack of oxygen being transported to tissues and organs. Since red blood cells are responsible for delivering oxygen, a reduced number of these cells means you don’t have enough oxygen in the body either.
Your anemia may be mild to severe. Symptoms of anemia include:
- dizziness
- fatigue
- irritability
- shortness of breath
- weakness
Anemia can also cause you to pass out. Severe cases can lead to widespread organ damage, which can be fatal.
Thalassemia and genetics
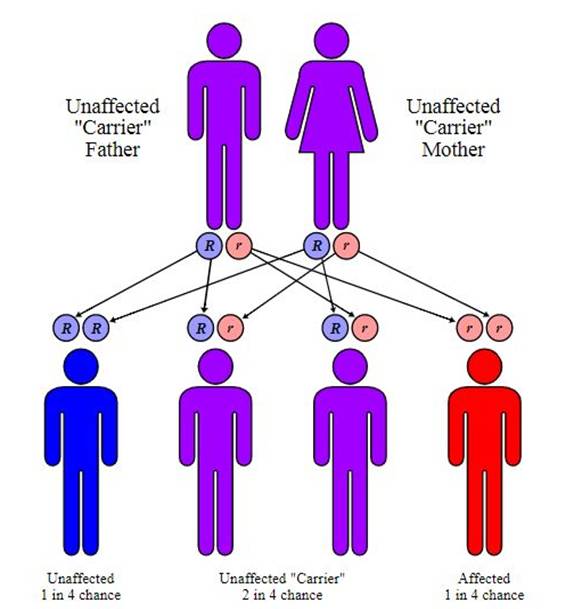
Thalassemia is genetic in nature. To develop full thalassemia, both of your parents must be carriers of the disease. As a result, you will have two mutated genes.
It’s also possible to become a carrier of thalassemia, where you only have one mutated gene and not two from both parents. Either one or both of your parents must have the condition or be a carrier of it. This means that you inherit one mutated gene from either one of your parents.
It’s important to get tested if one of your parents or a relative has some form of the disease.
Thalassemia minor
In alpha minor cases, two genes are missing. In beta minor, one gene is missing. People with thalassemia minor don’t usually have any symptoms. If they do, it’s likely to be minor anemia. The condition is classified as either alpha or beta thalassemia minor.
Even if thalassemia minor doesn’t cause any noticeable symptoms, you can still be a carrier for the disease. This means that, if you have children, they could develop some form of the gene mutation.
SYMPTOMS
There are several types of thalassemia. The signs and symptoms you have depend on the type and severity of your condition.
Thalassemia signs and symptoms can include:
· Fatigue
· Weakness
· Pale or yellowish skin
· Facial bone deformities
· Slow growth
· Abdominal swelling
· Dark urine
Some babies show signs and symptoms of thalassemia at birth; others develop them during the first two years of life. Some people who have only one affected hemoglobin gene don't have thalassemia symptoms.
CAUSES
Thalassemia is caused by mutations in the DNA of cells that make hemoglobin — the substance in red blood cells that carries oxygen throughout your body. The mutations associated with thalassemia are passed from parents to children.
Hemoglobin molecules are made of chains called alpha and beta chains that can be affected by mutations. In thalassemia, the production of either the alpha or beta chains are reduced, resulting in either alpha-thalassemia or beta-thalassemia.
In alpha-thalassemia, the severity of thalassemia you have depends on the number of gene mutations you inherit from your parents. The more mutated genes, the more severe your thalassemia.
In beta-thalassemia, the severity of thalassemia you have depends on which part of the hemoglobin molecule is affected.
Alpha-thalassemia
Four genes are involved in making the alpha hemoglobin chain. You get two from each of your parents. If you inherit:
· One mutated gene, you'll have no signs or symptoms of thalassemia. But you are a carrier of the disease and can pass it on to your children.
· Two mutated genes, your thalassemia signs and symptoms will be mild. This condition might be called alpha-thalassemia trait.
· Three mutated genes, your signs and symptoms will be moderate to severe.
Inheriting four mutated genes is rare and usually results in stillbirth. Babies born with this condition often die shortly after birth or require lifelong transfusion therapy. In rare cases, a child born with this condition can be treated with transfusions and a stem cell transplant.
Beta-thalassemia
Two genes are involved in making the beta hemoglobin chain. You get one from each of your parents. If you inherit:
· One mutated gene, you'll have mild signs and symptoms. This condition is called thalassemia minor or beta-thalassemia.
· Two mutated genes, your signs and symptoms will be moderate to severe. This condition is called thalassemia major, or Cooley anemia.
Babies born with two defective beta hemoglobin genes usually are healthy at birth but develop signs and symptoms within the first two years of life. A milder form, called thalassemia intermedia, also can result from two mutated genes.
RISK FACTORS
Factors that increase your risk of thalassemia include:
· Family history of thalassemia. Thalassemia is passed from parents to children through mutated hemoglobin genes.
· Certain ancestry. Thalassemia occurs most often in African Americans and in people of Mediterranean and Southeast Asian descent.
DIAGNOSIS
Presence of mutated gene that causes thalassemia can be easily detected with a special blood test called hemoglobin electrophoresis. This test can reveal whether a person is thalassemia carrier or not. This test separates out the different molecules in the red blood cells, allowing them to identify the abnormal type.
Depending on the type and severity of the thalassemia, a physical examination might also help your doctor make a diagnosis. For example, a severely enlarged spleen might suggest to your doctor that you have hemoglobin H disease.
TREATMENT
Treatments for thalassemias depend on the type and severity of the disorder. People who are carriers or who have alpha or beta thalassemia trait have mild or no symptoms. They’ll likely need little or no treatment.
Doctors use three standard treatments for moderate and severe forms of thalassemia. These treatments include blood transfusions, iron chelation (ke-LAY-shun) therapy, and folic acid supplements. Other treatments have been developed or are being tested, but they're used much less often.
Standard Treatments
Blood Transfusions
Transfusions of red blood cells are the main treatment for people who have moderate or severe thalassemias. This treatment gives you healthy red blood cells with normal hemoglobin.
During a blood transfusion, a needle is used to insert an intravenous (IV) line into one of your blood vessels. Through this line, you receive healthy blood. The procedure usually takes 1 to 4 hours.
Red blood cells live only for about 120 days. So, you may need repeated transfusions to maintain a healthy supply of red blood cells.
If you have hemoglobin H disease or beta thalassemia intermedia, you may need blood transfusions on occasion. For example, you may have transfusions when you have an infection or other illness, or when your anemia is severe enough to cause tiredness.
If you have beta thalassemia major (Cooley's anemia), you’ll likely need regular blood transfusions (often every 2 to 4 weeks). These transfusions will help you maintain normal hemoglobin and red blood cell levels.
Blood transfusions allow you to feel better, enjoy normal activities, and live into adulthood. This treatment is lifesaving, but it's expensive and carries a risk of transmitting infections and viruses (for example, hepatitis). However, the risk is very low in the United States because of careful blood screening.
Iron Chelation Therapy
The hemoglobin in red blood cells is an iron-rich protein. Thus, regular blood transfusions can lead to a buildup of iron in the blood. This condition is called iron overload. It damages the liver, heart, and other parts of the body.
To prevent this damage, doctors use iron chelation therapy to remove excess iron from the body. Two medicines are used for iron chelation therapy.
· Deferoxamine is a liquid medicine that's given slowly under the skin, usually with a small portable pump used overnight. This therapy takes time and can be mildly painful. Side effects include problems with vision and hearing.
· Deferasirox is a pill taken once daily. Side effects include headache, nausea (feeling sick to the stomach), vomiting, diarrhea, joint pain, and tiredness.
Folic Acid Supplements
Folic acid is a B vitamin that helps build healthy red blood cells. Your doctor may recommend folic acid supplements in addition to treatment with blood transfusions and/or iron chelation therapy.
Other Treatments
Other treatments for thalassemias have been developed or are being tested, but they're used much less often.
Blood and Marrow Stem Cell Transplant
A blood and marrow stem cell transplant replaces faulty stem cells with healthy ones from another person (a donor). Stem cells are the cells inside bone marrow that make red blood cells and other types of blood cells.
A stem cell transplant is the only treatment that can cure thalassemia. But only a small number of people who have severe thalassemias are able to find a good donor match and have the risky procedure.